Proceso de Simulación de Dinámica Molecular
La MDS es una técnica versátil que abarca varios tipos específicos de simulaciones, cada una adaptada para diferentes aplicaciones y escalas de tiempo/espacio. Dinámica Molecular Clásica o all atoms y de grano grueso o Coarse-Grained son los más utilizados, la diferencia radica en que all atoms es ideal cuando se requiere un alto nivel de detalle y precisión, mientras que el modelo coarse-grained es útil para simulaciones de sistemas extensos o para explorar fenómenos a escalas temporales y espaciales mayores, sacrificando parte de la resolución atómica.
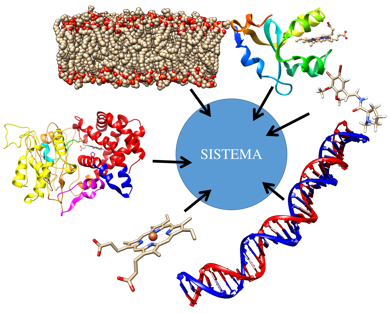
El proceso de MDS inicia con la Preparación del Sistema de Simulación: implica definir la estructura química del sistema, que se representa en términos de moléculas individuales y "monómeros" para polímeros biológicos, como proteínas, ácidos nucleicos, lípidos, etc.
La siguiente fase de la simulación de dinámica molecular es la creación de un archivo de topología para la estructura de la proteína. Con el software GROMAS se usa el programa pdb2gmx que genera una topología para el archivo de coordenadas de entrada.
Ahora es posible realizar estudios de dinámica molecular de sistemas biológicos complejos y de gran tamaño. Estos sistemas, por lo general, no están disponibles experimentalmente y se construyen mediante el ensamblaje manual de sus diferentes componentes.
La siguiente fase es colocar la estructura de la proteína en una caja de solvente particular para que pueda simularse en el entorno del mundo real. Después de crear una caja de solvente para la simulación, la siguiente fase en la simulación de dinámica molecular es realizar la solvatación. Es necesario un modelo de solvatación para simular mejor el entorno biológico del sistema. Hay muchas formas de solvatar un sistema molecular, con o sin condiciones de contorno. Por ejemplo, el sistema puede colocarse en una caja de agua dentro del marco de condiciones de contorno periódicas.
Posteriormtente, se debe neutralizar el sistema con contraiones que equilibren la carga a la solución. Por ejemplo, si tenemos una proteína con residuos ácidos cargados o con residuos básicos cargados, se pueden usar contraiones como Ca+, Na+, Cl- y colocarlos en posiciones idealizadas (mínimos de energía) para equilibrar las cargas correspondientes. No todos los residuos cargados requieren un contraión, ya que estos pueden estar involucrados en interacciones de puentes salinos.


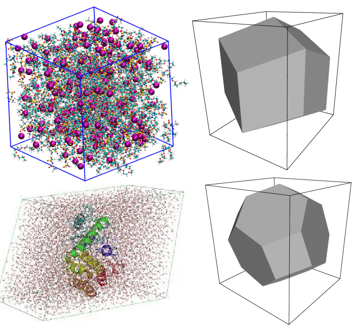
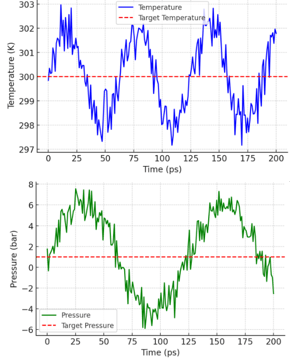
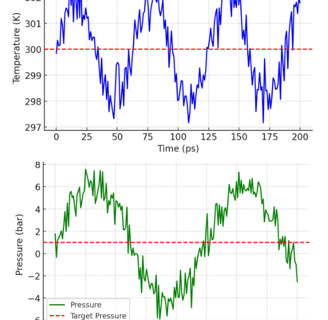
Una vez que se ha minimizado la energía de la estructura de la proteína, el siguiente paso es equilibrar la estructura para optimizarl y llevar el solvente a la temperatura en la que realizará el análisis de simulación, a esto se le llama ensamble canónico o en donde el número de moles, volumen y temperatura es constante (NVT). Posteriormente, sigue aplicar presión para crear una densidad dentro de la estructura de la proteína para que podamos realizar el análisis de simulación en esa región, llamado ensamble isobárico-isotérmico (NPT). El equilibrio de las proteínas se debe realizar en dos fases consecutivas.
Una vez que el sistema se ha equilibrado, está listo para las ejecuciones de producción. Se pueden generar múltiples trayectorias MD a partir de la configuración inicial (equilibrada). Además, el muestreo de diferentes estados iniciales mediante la elección de geometrías iniciales alternativas y procedimientos de equilibrio puede ayudar a lograr un mejor muestreo y convergencia de las simulaciones en general.
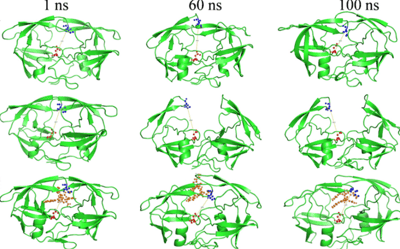

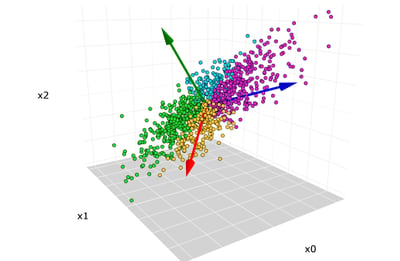
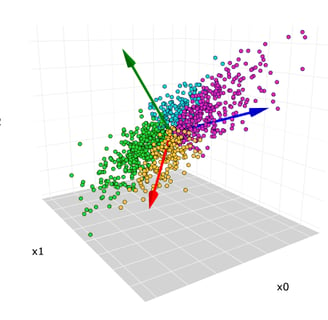
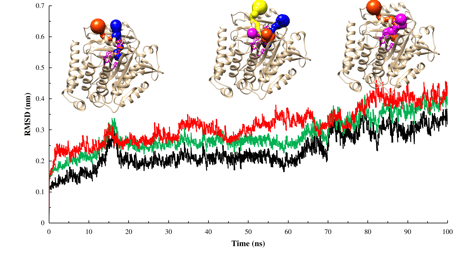
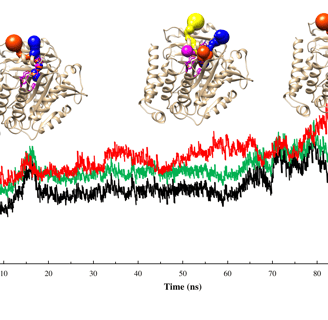
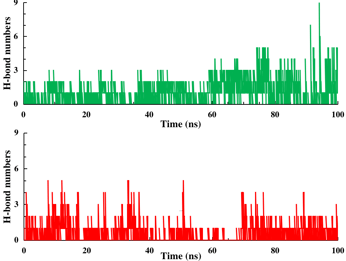
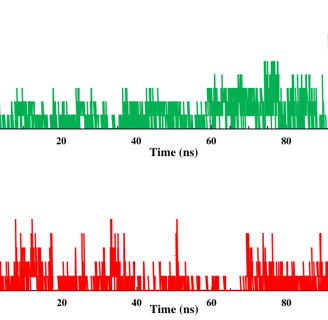
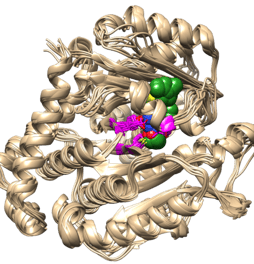
Para finalizar, se analiza los resultados de la MDS, primero se calcula RMSD para medir la distancia promedio entre un grupo de átomos (átomos de la estructura principal de las proteínas). Luego el RMSF que mide la desviación promedio de una partícula (un residuo de proteína) a lo largo del tiempo desde una posición de referencia (normalmente la posición promediada en el tiempo de la partícula). Posteriormente, se realiza el análisis de componentes principales (PCA), que convierte un conjunto de observaciones correlacionadas (movimiento de átomos en la proteína) en un conjunto de componentes principales que son linealmente independientes. Para calcular la energía libre de unión se usa MM-PBSA. También, se realiza el análisis de puentes de hidrógeno, entre otros análisis.
Consulta en línea
Complete este formulario y uno de nuestros expertos le responderá dentro de un día hábil.