Simulación de Dinámica Molecular
Alquimista Molecular ofrece servicios simulaciones de dinámica molecular, estas son técnicas computacionales que simulan el comportamiento de las moléculas a lo largo del tiempo. Al rastrear las posiciones y velocidades de los átomos individuales dentro de un sistema, se obtendran información sobre cómo estas moléculas interactúan, se pliegan y funcionan.
Ofrecemos los servicios de MDS de all-atoms, la cual es una técnica que permite estudiar el comportamiento de sistemas biológicos a nivel atómico a lo largo del tiempo. En este enfoque, cada átomo de las biomoléculas y su entorno (agua, iones, membranas) se modela explícitamente, permitiendo una descripción detallada de las interacciones moleculares.
Precisión en la Representación Molecular: captura interacciones atómicas específicas como enlaces de hidrógeno, fuerzas de van der Waals y efectos electrostáticos con alta resolución.
Estudio de la Estabilidad y Dinámica Estructural: Permite analizar cómo cambian la conformación y flexibilidad de proteínas, ADN, ARN y complejos biomoleculares bajo diferentes condiciones.
Diseño de Fármacos y Modelado de Interacciones: Es clave en la predicción del reconocimiento molecular entre proteínas y ligandos, ayudando a optimizar compuestos con potencial terapéutico.
Condiciones Realistas: Se pueden simular ambientes celulares específicos (pH, temperatura, presión) para entender procesos biológicos en condiciones cercanas a la realidad.
Análisis de Procesos Biológicos Dinámicos: Se usa para estudiar plegamiento de proteínas, mecanismos de resistencia a fármacos, transporte de moléculas y función enzimática.
¿Qué ofrecemos?
Simulación de Dinámica Molecular de Proteína
Simulación de Dinámica Molecular de Proteína-Ligando
Simulación de Dinámica Molecular de Proteína-Proteína
Simulación de Dinámica Molecular de Proteína-DNA o RNA
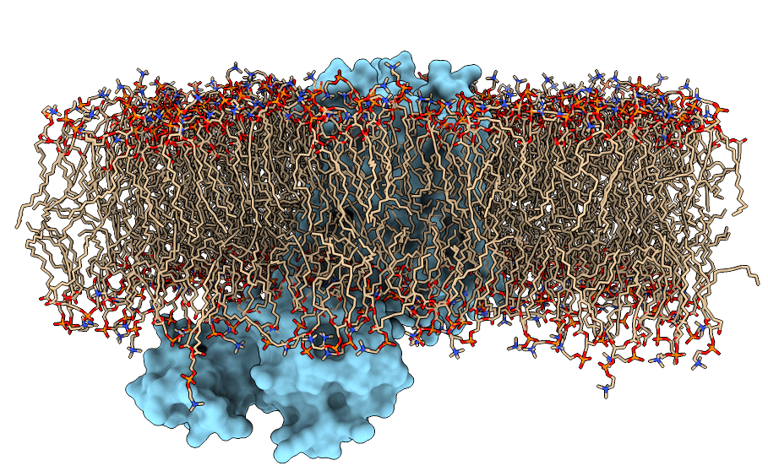
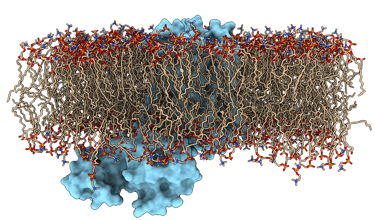
Simulación de Dinámica Molecular de Proteína-Lípido
Simulación de Dinámica Molecular de Proteína en Membrana
Simulación de Dinámica Molecular de Proteínas (Mutaciones)
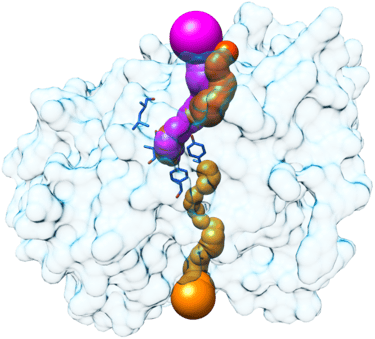
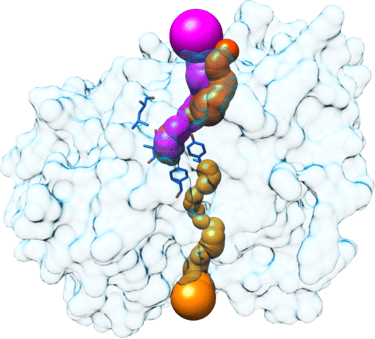
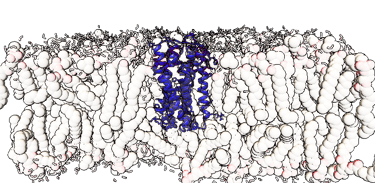
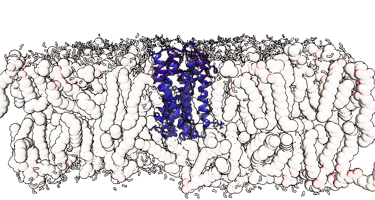
Métodos de análisis
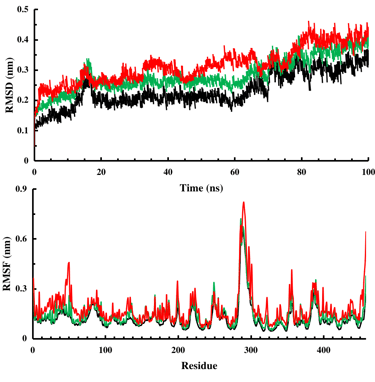
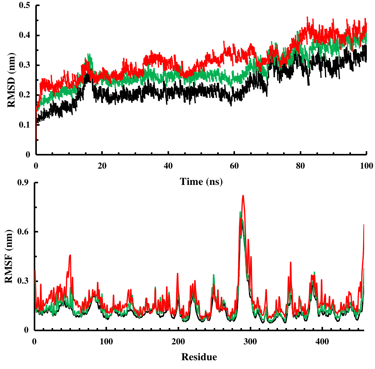
RMSD (root-mean-square-deviation)
RMSF (root-mean-square fluctuation)
Radio de giro (Rg):
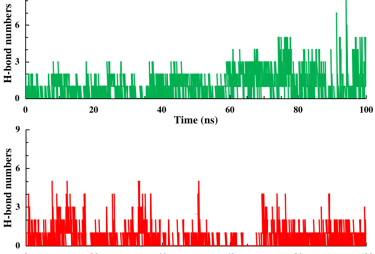
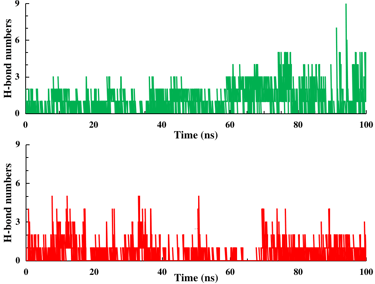
Análisis de Puentes de Hidrógeno
SASA (Área de Superficie Accesible al Solvente)
Análisis de frecuencia de contacto
Análisis de área de contacto
MM/PBSA & MMPBSA Energía Libre de Unón
Análisis de Componentes Principales (PCA)
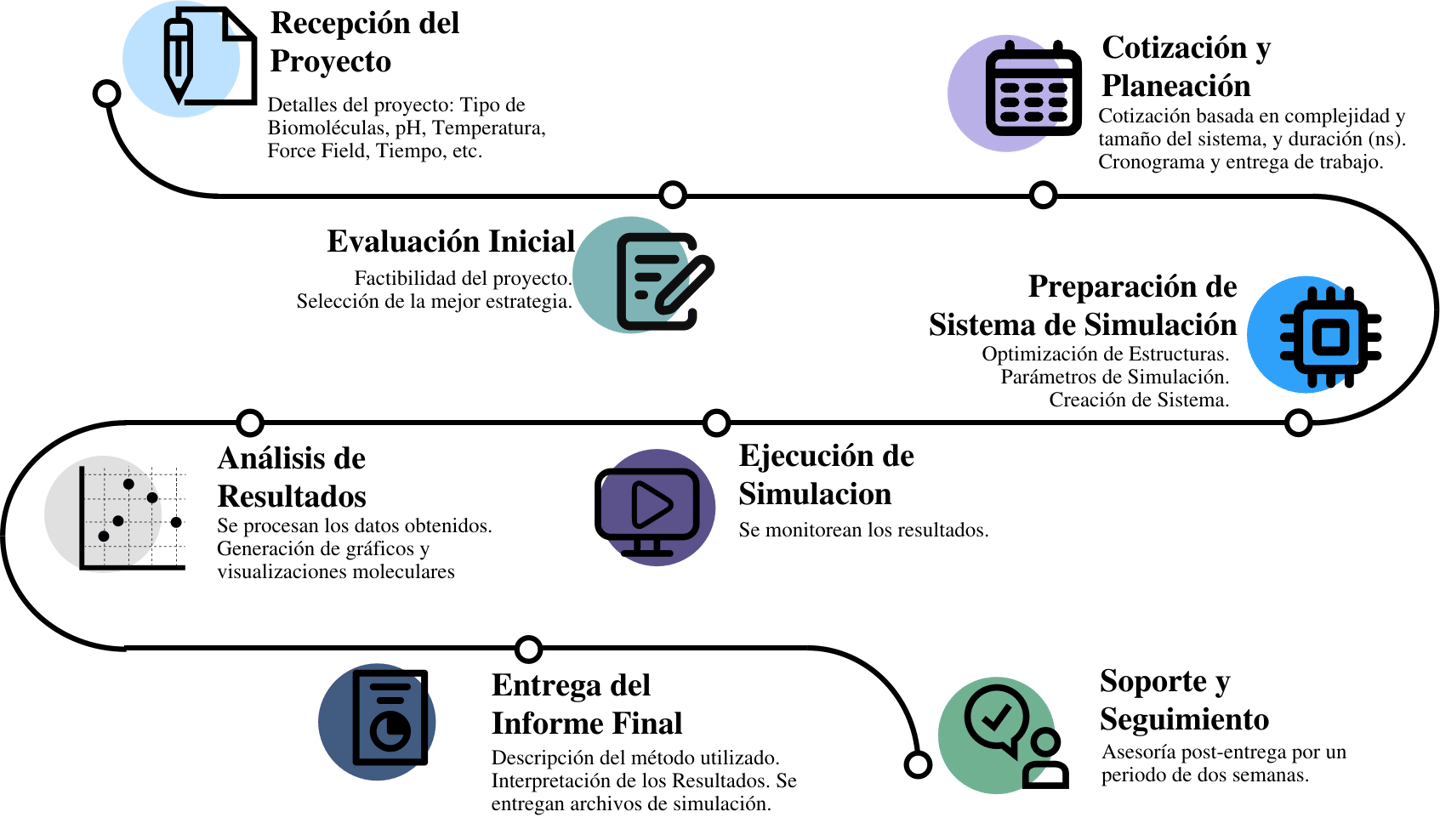
Obtenga sus resultados dentro de 48-72h
Dependiendo de la complejidad y tamaño del sistema, y duración de simulación (ns)
Consulta en línea
Complete este formulario y uno de nuestros expertos le responderá dentro de un día hábil.